
New knowledge about muscular dystrophy
Researchers at Aarhus University have revealed a previously unknown function of a cellular enzyme that can disperse toxic aggregates in the cells of patients with muscular dystrophy.

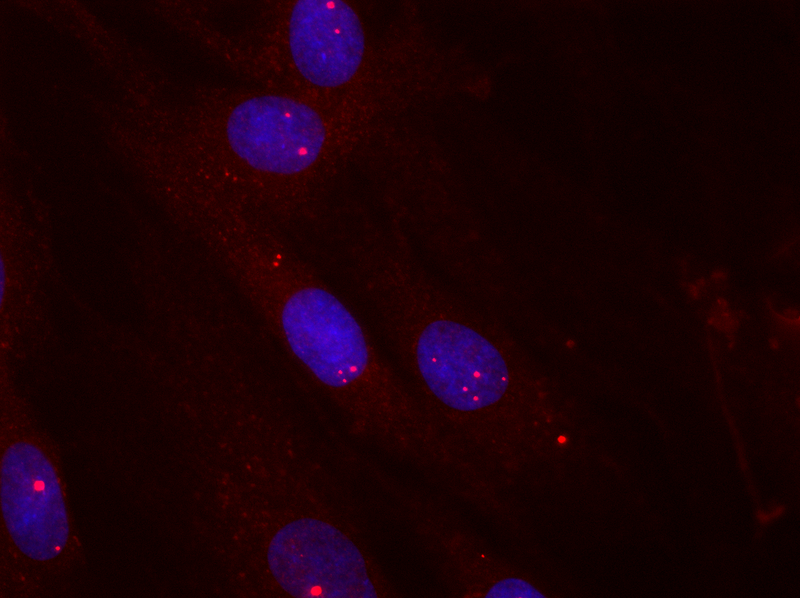
The most common form of muscular dystrophy among adults is dystrophia myotonica type 1 (DM1), where approximately 1 in every 8000 is affected by the disease. The severity of the disease varies from mild forms to severe congenital forms. It is dominantly inherited and accumulates through generations, gaining increased severity and lowered age of onset. DM1 is characterised by accumulating toxic aggregates of ribonucleic acids (RNA) from a specific mutated gene (see figure 1).
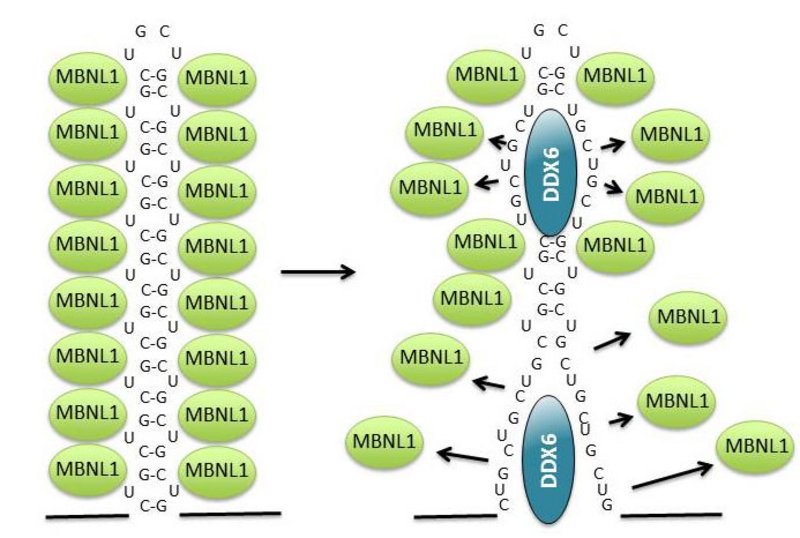
When this RNA, which contains thousands of CUG nucleotide repeats, builds up in the cell, it attracts several cellular proteins, including muscleblind 1 (MBNL1) (see figure 2, left). This binding inhibits the normal function of MBNL1, which means that the cellular level of a number of specific proteins becomes deregulated and the disease develops.
Enzyme characterisation in muscular dystrophy patients
The researchers work at the Department of Molecular Biology and Genetics and the Department of Biomedicine, Aarhus University, where they characterised an enzyme called DDX6, found in both normal cells and cells from muscular dystrophy patients.
The enzyme constantly tries to disperse the toxic aggregates and release MBNL1 in cells from muscular dystrophy patients, which means that the protein can carry out its normal function (see figure 2, right). The enzyme is found in many different cell types, where it performs a number of vital functions. The researchers showed that an artificial increase in the level of DDX6 in muscular dystrophy cells reduces the number of RNA aggregates, while more are formed when DDX6 is removed from the cells.
DDX6 belongs to a class of enzymes called helicases, which can change RNA structure and also regulate the ability of proteins to bind RNA. By purifying DDX6 from human cells, the researchers succeeded in getting the enzyme to bind and carry out an enzymatic reaction outside the cell, thus changing the structure of the toxic RNA.
These results indicate that DDX6 has a direct impact on toxic RNA aggregation in cells from muscular dystrophy patients in addition to its normal functions.
It remains unlikely that DDX6 can be used directly in the treatment of muscular dystrophy, since the enzyme carries out a number of important processes in the cell, which could potentially become deregulated leading to other diseases. However, the results provide important insights into the basic mechanisms of the disease, and natural differences in enzyme levels in different types of cells (or individuals) could possibly explain observed tissue-specific differences in the development of the disease.
The research team consists of Olof Pettersson, Lars Aagaard and Thomas G. Jensen, Department of Biomedicine, and Rune Thomsen, Diana Andrejeva (presently at University of Copenhagen) and Christian Kroun Damgaard, Department of Molecular Biology and Genetics, Aarhus University.
Link to the scientific article in the international journal Nucleic Acids Research: DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1.
Contact
Associate Professor Christian K. Damgaard
Department of Molecular Biology and Genetics
Aarhus University, Denmark
cd@mb.au.dk - +45 2970 0599