Kasper Andreasen Clowes, Camilla Gottlieb Andersen, Adriana Chrenková & Bjørn Panyella Pedersen
Sugar transporters are essential for the long-distance carbon allocation in plants and contribute to both biotic and abiotic stress responses yet the molecular mechanisms of substrate specificity and conformational cycling are only beginning to emerge. Recent structural and biophysical studies of the Sugar Transport Protein, the Sucrose Carrier, and the Sugar Will Eventually be Exported Transporter families have revealed how both conserved and divergent features shape substrate recognition and conformational cycling. Despite these advances, complete transport cycles and native substrate-bound states remain unresolved for several transporters. Advancing our understanding will require integrating structural, dynamic, and functional insights to define the mechanisms that govern sugar transport which could enable future engineering of transport proteins relevant for plant resilience and productivity.
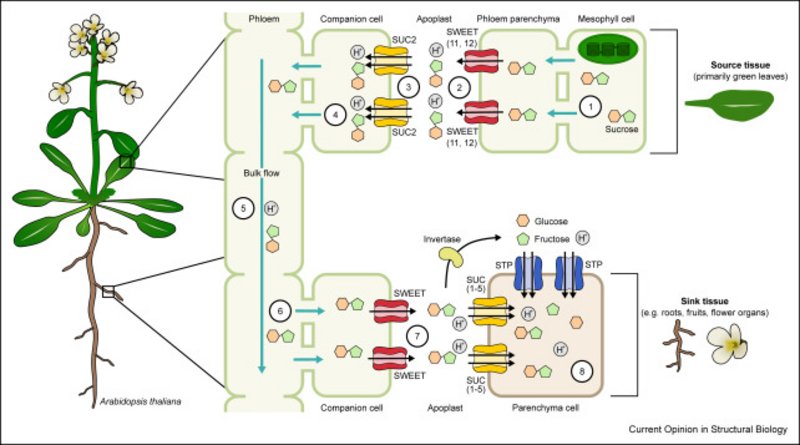
Figure: Transport of sucrose from source to sink tissues via the apoplastic pathway.
Lynette Nel, Jan H. Driller, Ronja Driller, Kelly M. Frain & Bjørn P. Pedersen
The organic cation choline is essential for eukaryotic metabolism. Recently, the feline leukemia virus subgroup C receptor–related (FLVCR, SLC49) family was demonstrated as central for basal choline transport, questioning the role of the choline transporter-like (CTL, SLC44) family in this capacity. Here, we use Xenopus laevis oocytes to confirm that FLVCR1 (SLC49A1) and FLVCR2 (SLC49A2) proteins are choline transporters. CTL1 (SLC44A1) does not transport choline under the same conditions, supported by other CTL proteins, Arabidopsis thaliana CherI and Saccharomyces cerevisiae PNS1, which also display no choline transport activity. We present the atomic structures of FLVCR2, CTL1, and PNS1. The 3.4 Å cryo-EM structure of FLVCR2 has choline in the binding pocket. The 3.3 Å cryo-EM structure of CTL1 and the 2.7 Å crystal structure of PNS1 reveal an unusual protein fold, weakly related to the mitochondrial carrier family (SLC25). The unusual fold appears incompatible with transmembrane transport and implies a different and, so far, unknown function for CTL proteins. Our results support FLVCR proteins as choline transporters and suggest a nontransport role for CTL proteins.
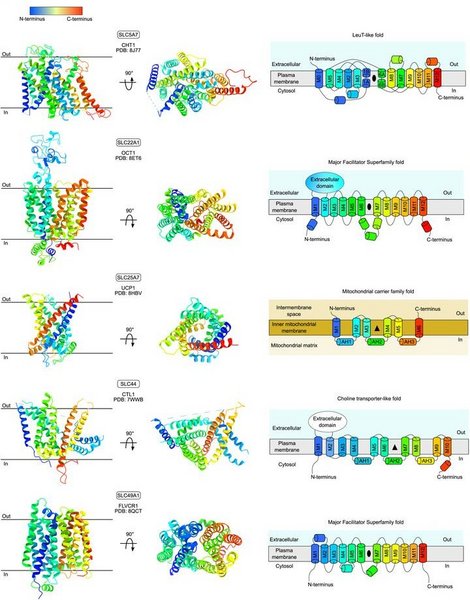
Figure: Experimentally determined structural overview of the transporters of choline and ethanolamine in humans.
Hassan Maklad, Tom Kache, Aurélie Roth, Maria Mamkaeva, Cédric Govaerts, Jelle Hendrix & Chloé Martens
Multidrug transporters are membrane proteins that can transport an ensemble of structurally dissimilar compounds and contribute to bacterial multidrug resistance (MDR) by exporting different antibiotics from the cell. However, whether they transport different substrates through a common mechanism or via distinct substrate-dependent mechanisms remains unclear. In this work, we used single-molecule Förster resonance energy transfer (smFRET) to measure time-resolved conformational dynamics of LmrP, a multidrug transporter of the Major Facilitator Superfamily (MFS). We present high-resolution conformational landscapes of LmrP in the presence of different antibiotics. Through multi-parameter Hidden Markov Modeling (mpH2MM), we uncovered transient states and quantified their sub-millisecond interconversion kinetics. We observed antibiotic-dependent heterogeneity in the conformational landscape, both in accessible states and in interconversion rates. Notably, poorly or non-transported antibiotics slow down transition kinetics, pointing to rapid state interconversion as a driver of efficient transport. This suggests that MFS MDR transporters bind and export structurally dissimilar antibiotics by relying on an array of underlying conformational states with ligand-dictated interconversion rates. This work provides novel insights into the mechanism of MDR transporters and advocates for combined structure/dynamics-based drug design when targeting their function.
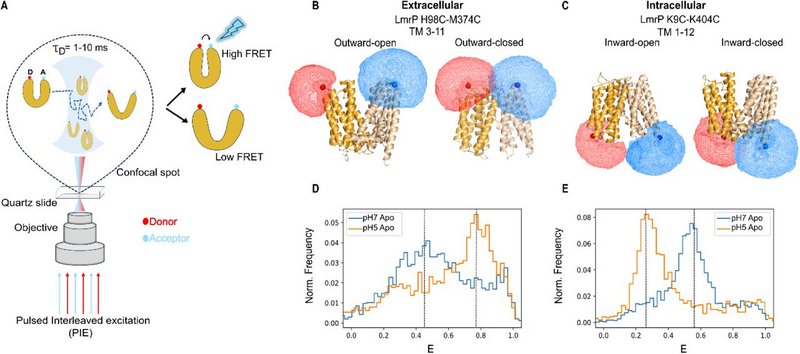
Figure: smFRET charts the conformational landscape of LmrP. A. Schematic illustration of the PIE smFRET experiment setup. B and C. Cartoon representation of LmrP helices with 98C (TM3)-374C (TM11) and LmrP 9C (TM1)-404C(TM12) in different conformations. D and E. Burst-wise FRET efficiency histograms showing the smFRET signatures of intracellular and extracellular distance reporter mutants.
Bjørn Lildal Amsinck, Ines Benhammouche, Adriana Chrenková, Alexander J.D. Snow & Bjørn Panyella Pedersen
Plant hormone transporters are essential for regulating phytohormone distribution at the cellular, tissue, and organ levels in plants. These proteins control hormone movement across membranes, shaping the spatial and temporal hormone gradients that underpin growth, development, and stress responses. This review surveys secondary active transporters implicated in the transport of auxin, cytokinin, abscisic acid, and gibberellins. We focus on five major transporter superfamilies: MFS, BART, APC, TOG, and DMT, highlighting their structural organization, transport mechanisms, and substrate specificity. Despite differences in substrate and regulation, members within each superfamily share conserved folds and mechanistic principles. Transport typically proceeds via alternating access models, including elevator, rocker-switch, and rocking-bundle mechanisms, which enable directional movement across chemically distinct environments. We show that structural classification offers an informative framework for predicting substrate interactions, energy coupling, and regulatory motifs, ultimately improving our understanding of hormone transport in plants. As more plant transporters are resolved structurally, this approach will be critical for uncovering how transport is coupled to energy sources and integrated into broader signaling networks.

Figure: Schematic representation of auxin, cytokinin, abscisic acid, and gibberellin secondary active transporters identified to date in Arabidopsis thaliana. Transporter localization at the plasma membrane or internal membranes, as well as the direction of transport (arrows), is indicated. Colors represent the transporter superfamilies and their corresponding protein folds, as indicated.
Nicolas S. Gonzalez-Foutel, Ankush Garg, Evi Setiani Lande, Assia Khalild, Line Mørkholt Lund, Victoria Birkedal, Chloé Martens & Magnus Kjaergaard
Biomolecular condensates form dynamic compartments that regulate biochemical reactions in cells. Condensates recruit many kinases and regulate their enzymatic activity. Condensates alter the rate of enzymatic reactions through several opposing effects, so it is unclear whether these mostly enhance or retard phosphorylation. Here, we use a synthetic condensate formed by intrinsically disordered proteins to show that slow diffusion in the condensate controls phosphorylation kinetics in the dense phase. We vary the length of substrates by appending phase-separating repeat proteins of different lengths, in order to study how phosphorylation depends on partitioning, diffusion and volume fraction across substrate motifs with different intrinsic kinetics. The condensate environment is generally inhibitory to phosphorylation, although the enzyme remains intact. This inhibition is partially offset by an enhanced reaction rate in the dilute phase, likely due to soluble nanoclusters. Phosphorylation rates are strongly correlated to diffusion coefficients of substrates in the condensate, suggesting mass-transport limitation. Our results suggest that condensates can modify the substrate usage of a kinase via different trade-offs between diffusion and partitioning. We suggest that diffusion limitations are likely a common feature of many macromolecular reactions in condensates, and that high fluidity is crucial for condensates to act as reaction crucibles.
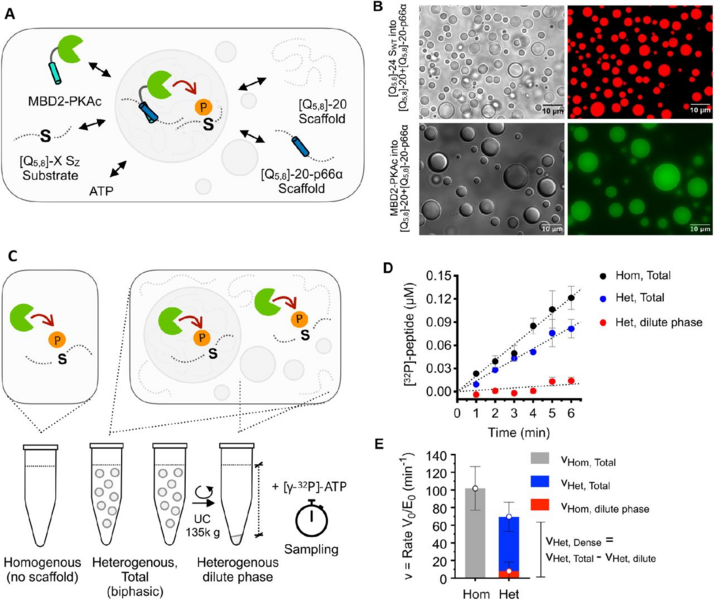
Figure: A) Schematic of condensates model system. B) DIC and confocal fluorescence microscopy of spherical condensates with fluorophore labelled substrate and enzyme. C) Schematic procedure for kinetic experiments. D) Time course of radioactive-P-phosphorylated substrate in homogeneous solution or different phase samples. E) Decomposition of phosphorylation reaction rates.
Adel Hussein, Xihui Zhang, Michael Schlame, Bjørn P. Pedersen & David L. Stokes
KdpFABC is a hetero-tetrameric potassium pump that uses ATP to import potassium and thereby maintain homeostasis in bacteria under stress conditions. KdpA is a channel-like subunit with a selectivity filter that binds potassium from the periplasm. K+ then moves through a ∼40Å-long intramembrane tunnel to reach a canonical binding site in KdpB. KdpB is a P-type ATPase that orchestrates conformational changes associated with the Post-Albers reaction cycle, involving E1 and E2 conformations and formation of an aspartyl phosphate intermediate as a way of coupling ATP hydrolysis to K+ transport. To elucidate the associated structural changes in a lipid environment, we reconstituted wild-type KdpFABC into lipid nanodiscs and used cryo-EM to image the complex under active turnover. The resulting six high resolution (2.1-2.7 Å) structures provide new insight into the sequence of allosteric changes that produce occlusion of K+ at the canonical binding site and expulsion of K+ from this site and into a low-affinity release site. The structures also reveal two types of lipids bound to the complex. Specifically, two structural lipids bind at subunit interfaces and ∼20 annular lipids are seen at the periphery of the complex. In addition, we tested functional effects of mutations to residues at the KdpA/KdpB interface. ATPase and transport assays were used to document functional defects that reflect delipidation of structurally compromised complexes. We conclude that lipids play an integral role in structure and function of the KdpFABC complex.
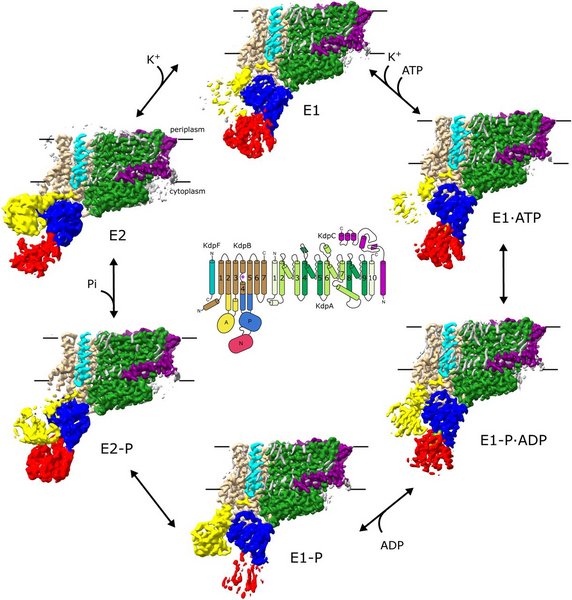
Figure: Cryo-EM structures of KdpFABC in lipid nanodiscs. Six density maps were derived from the images recorded under turnover conditions and assigned to a specific intermediate state in the Post-Albers reaction cycle. The diagram in the middle depicts helical topology. These density maps are hybrid structures with the membrane domains derived from conventional reconstruction with cryoSPARC and cytoplasmic domains with RELION. The membrane region is indicated by horizontal black lines.
Kien Lam Ung, Lukas Schulz, Lorena Zuzic, Bjørn Lildal Amsinck, Sarah Koutnik-Abele, Ines Benhammouche, Camilla Gottlieb Andersen, Lynette Nel, Birgit Schiøtt, David L. Stokes, Ulrich Zeno Hammes & Bjørn Panyella Pedersen
Auxins are plant hormones that direct the growth and development of organisms on the basis of environmental cues. Indole-3-acetic acid (IAA) is the most abundant auxin in most plants. A variety of membrane transport proteins work together to distribute auxins. These include the AUX/LAX protein family that mediate auxin import from the apoplast to the cytosol. Here we use structural and biophysical approaches combined with molecular dynamics to study transport by Arabidopsis thaliana LAX3, which is essential for plant root formation. Transport assays document high-affinity transport of IAA, as well as competitive behaviour of the synthetic phenoxyacetic acid auxin herbicide 2,4-dichlorophenoxyacetic acid and the auxin transport inhibitors 1-naphthoxyacetic acid and 2-naphthoxyacetic acid. Four cryo-EM structures were solved with resolutions of 2.9–3.4 Å: an inward open apo structure, two inward semi-occluded structures in complex with IAA and 2,4-dichlorophenoxyacetic acid, and a fully occluded structure in complex with 2-naphthoxyacetic acid. Structurally, LAX3 consists of a bundle and a scaffold domain. The ligand-binding site is sandwiched between these domains with two histidines occupying positions analogous to the sodium-binding sites in distantly related sodium:neurotransmitter transporters. This architecture suggests that these histidines couple transport to the proton motive force. Molecular dynamics simulations are used to explore substrate binding and release, including their dependence on specific protonation states. This study advances our understanding of auxin recognition and transport by AUX/LAX, providing insights into a fundamental aspect of plant physiology and development.
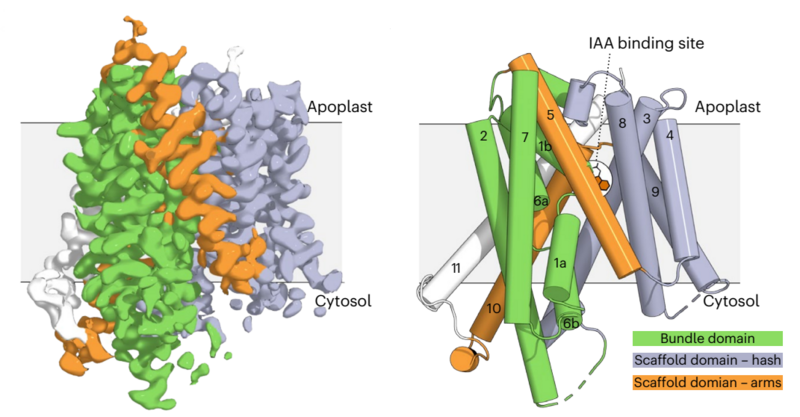
Figure: The cryo-EM electrostatic potential map of the 3.21 Å structure of LAX3 bound to IAA. The bundle domain (green) consisting of M1, M3, M6 and M8, and the scaffold domain (light purple) with M2, M4, M5, M7 and M9, form the core of the transporter. In addition, the transporter has two arms, M5 and M10 (orange), and an extra C-terminal helix, M11 (white), which breaks the internal symmetry of the transporter.
The Center for Active Transport of Plant Hormones (Plant-PATH) is a Center of Excellence funded by the Danish National Research Foundation.
