
Aggregation of proteins in cells may result in diseases
Changes in the structure of proteins can lead to various diseases, such as Alzheimer’s, type 2 diabetes and corneal dystrophy. A research team from Aarhus University has now discovered how a particular protein can damage cells. These results may lead to the development of drugs to treat corneal dystrophy in the future.

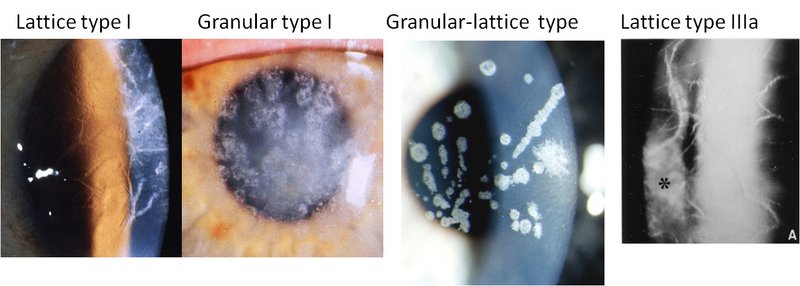
Many diseases are caused by proteins losing their natural three-dimensional structure and thus their function. In most cases, the damaged proteins are degraded by different systems in the cells, but in some cases, the proteins begin to aggregate and form very well-organised rope-like structures called fibrils. These structures have now been linked to many different diseases, such as Alzheimer’s, Parkinson’s, type 2 diabetes and corneal dystrophies (opaqueness in the cornea).
It has long been known that corneal dystrophy is caused by a mutation of a protein called TGFBIp in the cornea. This causes the protein to aggregate over a number of years, which finally makes the cornea opaque, eventually leading to blindness. Corneal dystrophy is hereditary, and there is currently no treatment for this disease. The only alternative is a corneal transplant, but even after a transplant, some patients experience a recurrence of the symptoms. It is therefore important to find a reason for this aggregation, and this is what the Aarhus researchers have just accomplished.
High amounts of TGFBIp protein are damaging
The research team studied the protein that causes corneal dystrophy, and they found that the mutation of the protein changes its stability. When the protein involved in corneal dystrophy is present in low amounts, the change from a protein with a natural structure to well-organised fibrils is a slow process, and it takes place via several different intermediate stages. In this case, the cell damage is minimal. However, when protein is present in larger quantities, the change from a protein with a natural structure to fibrils is very rapid and involves very few and well-defined intermediate stages. On the other hand, this leads to extensive cell damage.
With these studies, the researchers gained unique insight into how the amount of protein in solution can determine the mechanism underlying the formation of well-organised protein fibrils.
The results may explain why some diseases with a prevalence of fibrils result in cell death, while others do not. With this knowledge, it might be possible in the future to prevent this aggregation of damaging proteins by developing drugs for the treatment of corneal dystrophy.
The researchers behind the results, which have just been published in the international Journal of Biological Chemistry (JBC), are affiliated with the Danish National Research Foundation’s Centre for Insoluble Protein Structures (inSPIN) located at the Interdisciplinary Nanoscience Center (iNANO), Department of Chemistry and the Department of Molecular Biology and Genetics (MBG), Aarhus University, Denmark.
Link to the scientific article in JBC: Polymorphic fibrillation of the destabilized 4th fasciclin-1 domain mutant A546T of the transforming growth factor-? induced protein (TGFBIp) occurs through multiple pathways with different oligomeric intermediates
Maria Andreasen1, Søren B. Nielsen1, Kasper Runager1, Gunna Christiansen2, Niels Chr. Nielsen3, Jan J. Enghild1, Daniel E. Otzen1
1Center for Insoluble Protein Structures (inSPIN), Interdisciplinary Nanoscience Center (iNANO) and
Department of Molecular Biology and Genetics, Aarhus University, DK-8000 Aarhus, Denmark
2Department of Biomedicine, Aarhus University, DK-8000 Aarhus, Denmark,
3Center for Insoluble Protein Structures (inSPIN), Interdisciplinary Nanoscience Center (iNANO) and Department of Chemistry, Aarhus University, DK-8000 Aarhus, Denmark
Contact
Professor Daniel Otzen
Interdisciplinary Nanoscience Center (iNANO) and
Department of Molecular Biology and Genetics (MBG)
Aarhus University, Denmark
+45 2072 5238, dao@inano.au.dk
Text: Maria Andreasen and Lisbeth Heilesen
Translation into English: Lisbeth Heilesen
15 October 2012