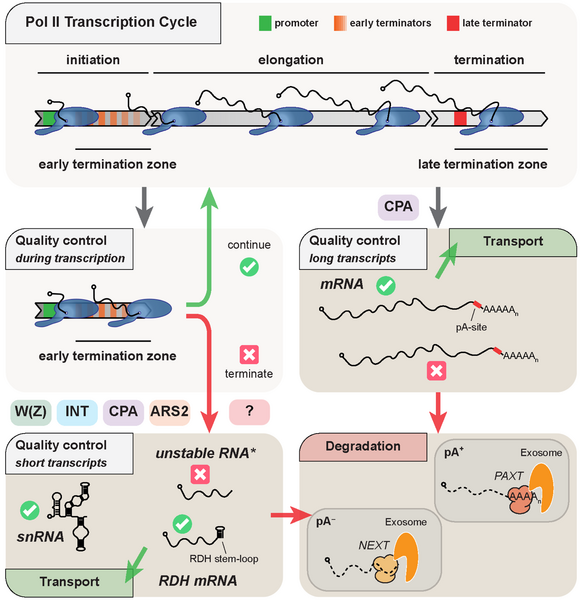
Canonical genes (encoding mRNA, tRNA, rRNA, sn(o)RNA, lncRNA) only represent a fraction of the mammalian genome, as most DNA is subject to Pol II transcription, producing e.g. enhancer (e)RNAs and promoter upstream transcripts (PROMPTs). The vast majority of these non-canonical transcription events are subject to early termination coupled to RNA decay. Premature termination also takes place at canonical transcription units with as much as 80% of transcription events ending prematurely in processes performed by different machineries.
Our laboratory aims to uncover early transcription termination mechanisms and their interplays with other nuclear processes such as RNA transport or degradation. To this end, we use multiple state-of-the-art transcriptome-wide sequencing techniques as well as high throughput mass spectrometry approaches, by which we have previously identified Integrator (Lykke-Andersen et al., 2021) and ARS2 (Andersen et al., 2013; Giacometti et al., 2017; Iasillo et al., 2017, Rouviere et al., 2023) as factors involved in early transcription termination. Our studies provide insights on the regulation of mammalian transcription as well as the physiological consequences arising from any defect in these control mechanisms.
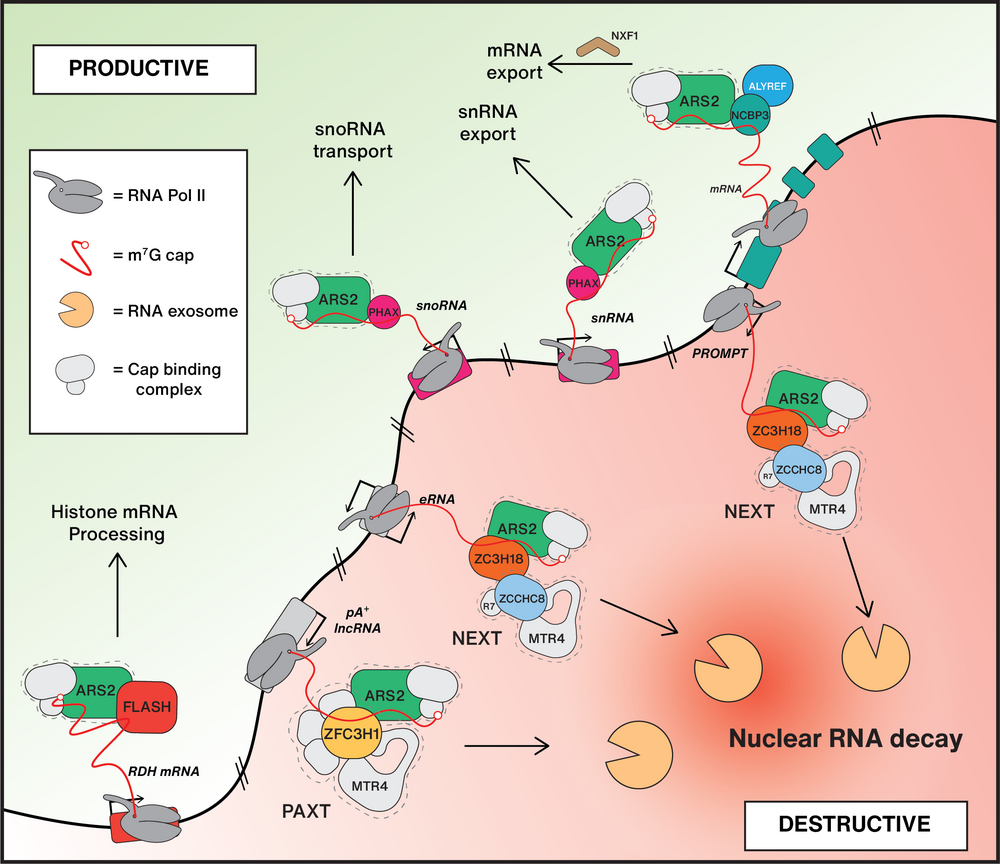
Being predominantly transcribed by Pol II, labile RNA species, like eRNAs and PROMPTs, share common features with their well-studied functional counterparts, snRNA, histone RNA and traditional mRNA, yet they meet a completely different cellular fate. This motivates a major question in our laboratory: How are functional transcripts sorted from the sea of futile transcription by-products that surrounds them?
To comprehend the logic underlying nuclear sorting of RNA at the post-transcriptional level, we aim to define and characterize key factors maintaining the necessary equilibrium between RNA biogenesis and destruction. We have pioneered characterization of the NEXT and PAXT adaptors of the ribonucleolytic RNA exosome (Lubas et al., 2011 and Meola et al., 2016), and described that both NEXT and PAXT can contact the cap-binding complex (CBC) and the ARS2 protein ubiquitously present at the 5’ends of nuclear Pol II-derived mRNA/ncRNA. These interactions are mutually exclusive with those of the CBC-ARS2 with the RNA transport factors PHAX, FLASH and ALY/REF, yielding a first glimpse of the competitive protein-protein connections, that we predict constitute the underlying molecular framework for the sorting of nuclear RNA.