The vast majority of biological processes ranging from signal transduction to gene expression depend on macromolecular interactions (protein-protein or protein-nucleic acid). We are studying the structural basis and functional relevance of macromolecular interactions involving bacterial translation elongation factors to obtain a better understanding of their roles in normal as well as in stressed or diseased states. Furthermore, we are investigating the possibilities of exploiting these interactions in drug development. Examples of potential projects are provided below.
EF-Tu belongs to the superfamily of G-proteins, which act as molecular switches for a regulatory purpose. Many diseases such as cancer and memory disorders are associated with the malfunctioning of G-proteins. The normal role of EF-Tu is to carry aminoacyl-tRNA (aa-tRNA) to the A-site of the mRNA-programmed ribosome in a GTP-dependent manner. Upon correct interaction between the codon exposed in the ribosomal A-site and the anticodon of the incoming aa-tRNA, GTP-hydrolysis is triggered. The resulting EF-Tu·GDP needs to be recycled by its guanine-nucleotide exchange factor, EF-Ts, which promotes the exchange of GDP with GTP thus enabling a new round in the elongation cycle to take place.
During its functional cycle, EF-Tu undergoes structural changes as illustrated by comparing the active, GTP-bound form and the inactive, GDP-bound form of the factor (Figure 1). We are exploring the dynamical aspects of these changes, using advanced single-molecule fluorescence microscopy based on fluorescence resonance energy transfer (FRET). In contrast to the general view of EF-Tu, we have shown that the conformational change of EF-Tu initiates on the ribosome, but is not completed until after dissociation. Furthermore, we have evidence that the structural change follows a pathway that cannot be predicted based on available structure models [1, 2].
We will produce fluorescently labelled EF-Tu variants to explore our findings in detail and gain information about the functional consequences of our results. We are using site-directed mutagenesis, protein expression and purification followed by protein labelling to produce EF-Tu variants suitable for our studies. The mutated and labelled EF-Tu’s are tested in functional assays prior to single-molecule fluorescence microscopy, which takes place in collaboration with Professor Yale E. Goldman at Pennsylvania University.

Figure 1. Comparison of the active (EF-Tu:GDPNP) and inactive (EF-Tu:GDP) conformations of EF-Tu.
In the inactive conformation, the three domains of EF-Tu form an open structure with a hole in the middle. Upon binding of GTP, domains 2 and 3 rotate by 90° relative to domain 1 causing closure of the hole. Also parts of domain I, switch region I (shown in magenta) and helix B, undergo structural rearrangements upon switching between the two conformations.
Viruses are obligate parasites that depend on host proteins and pathways during most steps of an infection. In particular, the replication of the genomes of RNA viruses has been found to require the "kidnapping" of host proteins by the virally encoded RNA-dependent RNA polymerase (RdRP; also known as replicase). Qb is a bacteriophage that infects E. coli (Figure 2). Upon entry of its (+)-stranded RNA genome into the host, the genome gets translated and the resulting RdRP forms a complex with EF-Tu, EF-Ts and ribosomal protein S1 (and eventually host factor, Hfq). We have solved the three-dimensional structure of this complex using X-ray crystallography [3]. The structure has revealed features of potential importance during genome replication i.e. in template binding, initiation, elongation and unwinding of the helix between template and product. We are subjecting the relevant amino acid residues or structural elements to structure-function analysis (see figure 3).
Our structure indicates that disruption of the interactions between the viral RdRP and host proteins may be a viable strategy in combating a viral infection. We will explore this possibility by developing a selection system that allows the identification of inhibitors of viral infection. Potential inhibitors will be selected from a library of peptide aptamers. Next, the mechanisms of the isolated inhibitors will be explored in binding- and activity assays.
The replication mechanism of Qb resembles the strategy applied by pathogenic viruses such as hepatitis C virus and polio virus. Thus, new information about the molecular details of replication of the Qb genome may be directly applicable to other (+)-stranded RNA viruses of medical relevance and lead to the identification of novel drug targets.
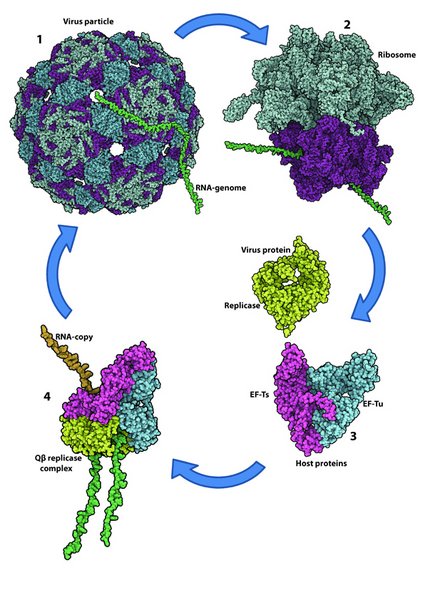
Figure 2. Structural illustration of the Qb infection cycle.
The virus particle binds to the surface of a host cell and injects the RNA genome (green) into the cytoplasm of the bacterium (1). Host ribosomes translate the (+)-stranded Qb genome into protein (2) resulting in the production of the viral replicase subunit (the RdRP; lime). The replicase subunit forms a complex with the host proteins EF-Tu (blue) and EF-Ts (magenta) resulting in an active Qb replicase complex capable of copying the RNA genome (3). The cycle is completed when the genome copies are packaged into coat proteins to form virions.
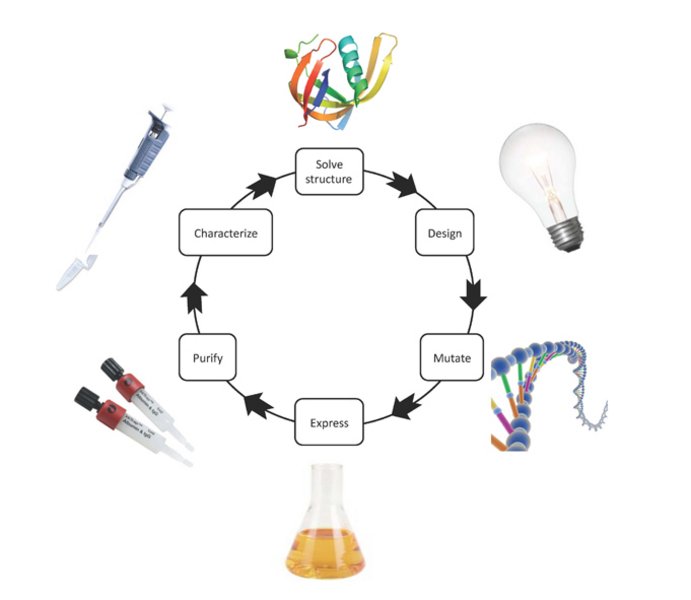
Figure 3. Flow of structure-function studies.
The three-dimensional structure of the studied protein forms the basis for designing mutations, which are predicted to affect the functional property of interest. The mutation is introduced into the protein-coding gene by site-directed mutagenesis. Subsequently, the mutant protein is expressed and purified. Finally, a functional characterization is carried out to test the working hypothesis.
There are approximately 7000 known genetically transmitted disorders in humans. 12% of these are the result of nonsense mutations, which cause the production of shortened, inactive protein products that may lead to disease.
Efforts have been made to develop therapeutic strategies to counteract the negative effects of premature termination codons resulting from nonsense mutations. One approach relies on enhancing the natural phenomenon of termination codon suppression through the translational readthrough mechanism (Figure 4). Termination of translation is based on the match between a stop codon in the ribosomal A site and a release factor. A near-cognate tRNA with an anticodon matching two out of the three positions of the stop codon can outcompete the release factor and cause the incorporation of its amino acid instead of termination. Therefore, the termination process is not 100% efficient and can be modulated via mutations or drugs.
Drugs that can potentially suppress premature termination of translation have been identified, but often suffer from severe toxicity problems. This indicates that drugging of premature termination is a viable strategy, but new methods of drug identification are required.
We will design a selection protocol that enables the identification of DNA-encoded, circular peptides that suppress premature termination of translation without disturbing other cellular functions. If successful, the selected circular peptides can restore the production of the full-length protein encoded by the mutated gene and thereby cure or relieve patients suffering from genetic diseases caused by nonsense mutations.
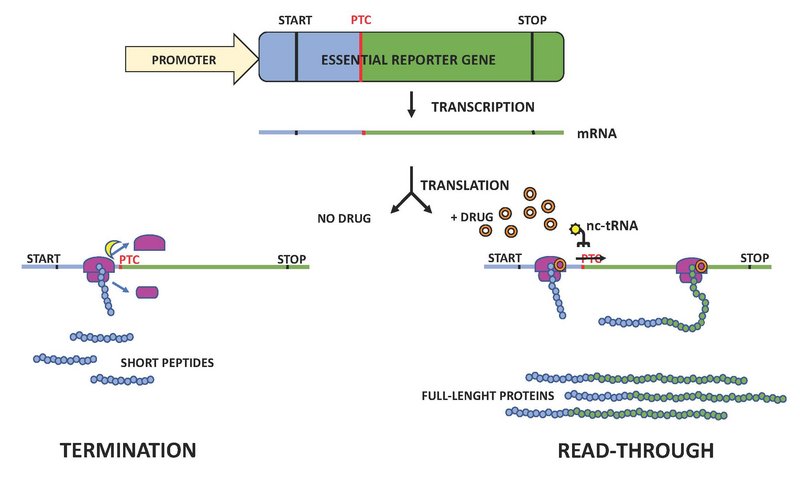
Figure 4. Suppression of translation termination.
A premature termination codon (PTC) is introduced in an essential reporter gene by site-directed mutagenesis. Without addition of a nonsense-suppression drug (orange circle), a release factor (yellow ”moon”) triggers premature termination resulting in ribosome (pink) dissociation and the release of a short peptide (left). In the presence of a nonsense-suppression drug (right), a near-cognate tRNA (nc-tRNA) can outcompete the release factor and cause the incorporation of the amino acid (yellow “sun”) transported by the nc-tRNA rather than termination. This results in the restoration of the full-length protein encoded by the reporter gene.